Читайте также:
|
Исходная структура молекулы ТРТ, взятая из PDB банка данных (Protein Data Bank [30], PDB код 1k4t) и затем дополнительно прооптимизирована по энергии в программе Gaussian 03W методом DFT(B3LYP) в базисе 6-31G*. Результирующие структуры молекулы ТРТ в лактонной и карбоксилатной формах.
Поскольку в литературе отсутствуют заряды на атомах молекулы ТРТ, нами были получены значения парциальных зарядов на атомах молекулы топотекана в обеих формах на основании неэмпирических (HF, DFT(B3LYP)) приближений уравнения Шредингера и 5 стандартных базисов (STO-3G, 3-21G, 6-31G, 6-31G*, 6-31G**) для получения электронной плотности, используемой в качестве входных данных при расчете атомных зарядов. Расчет атомных зарядов проводился с использованием двух групп методов, основанных на анализе Метода Хартри-Фока (HF) и Теории функционала плотности (DFT).
Метод Хартри — Фока — в квантовой механике приближённый метод решения уравнения Шрёдингера путём сведения многочастичной задачи к одночастичной в предположении, что каждая частица двигается в некотором усреднённом самосогласованном поле, создаваемом всеми остальными частицами системы [30].
Теория функционала плотности (англ. density functional theory, DFT) — метод расчёта электронной структуры систем многих частиц в квантовой физике и квантовой химии. В частности, применяется для расчёта электронной структуры молекул и конденсированного вещества [30].
Наиболее часто используются базисы STO-nG, где n — целое число, обозначающее количество простых гауссовых функций включёных в одну базисную функцию. В этих базисах одинаковое число гауссовых функций описывает основные и валентные орбитали. Такие базисы дают весьма грубые результаты, недостаточные для серьёзных научных расчетов, однако расчеты с применением таких базисов осуществляются значительно быстрее, нежели расчёты с более полными базисами [31].
Название валентно-расщеплённых базисных наборов, созданных группой Джона Поупла, выглядит обычно как X-YZg. Здесь X обозначает количество простых гауссовых функций, входящих в состав базисной функции атомной орбитали. Y и Z показывают, что валентные орбитали состоят из двух базисных функций каждая: первая из которых представляет собой линейную комбинацию Y простых гауссовых функций, а вторая — Z простых гауссовых функций. То есть две цифры после дефиса подразумевают, что данный базис является валентно-расщепленным, double-zeta. Если после дефиса стоит три или четыре цифры, то базис, соответственно, будет triple-, quadruple-zeta. В нашей работе используеться следующие базисы Попла: 3-21G, 6-31G, 6-31G*, 6-31G** [30-32].
В работе проводился анализ влияния трех основных факторов: Уровень теории (Фактор 1), Базисный набор волновых функций (Фактор 2) и Метод расчета заряда (Фактор 3) на расчетные значения атомных зарядов. Распределение зарядов на молекуле ТРТ наиболее сильно зависит от базиса (фактор 2), что также характерно и для более простых органических молекул [28-30]. Среди суб-факторов фактора 2 базис STO-3G в среднем дает заниженное значение заряда. Напротив, базисы 3-21G, 6-31G, 6-31G*, 6-31G** дают в среднем близкие результаты, что, в принципе, дает основание использовать эти базисы в равной степени для расчетов атомных зарядов ароматических молекул.
Базис 6-31G** является наиболее полным базисом, а базис 6-31G* дает аналогичное распределение зарядов. Из этого следует, что базис 6-31G* можно всегда использовать для расчета зарядов на ТРТ, вместо требующего значительно больших временных затрат базиса 6-31G**. Этот результат согласуется с мнением о том, что, начиная с базиса 6-31G* разброс значений зарядов слабо зависит от выбора базиса [29].
Неэмпирические методы расчета дают наиболее надежные результаты, поэтому полуэмпирические методы расчета в данной работе не использовались. В тоже время метод HF приводит в среднем к завышенным значениям атомных зарядов, и, таким образом, не может быть рекомендован для расчетов. Отличие результатов расчетов по методу HF от DFT(B3LYP) также отмечалось ранее для более простых молекул [28,29] и других ароматических молекул [29].
Таким образом, рекомендуемым методом расчета парциальных атомных зарядов на молекуле ТРТ являются методы МК или CHelpG на уровне теории DFT(B3LYP) в базисе 6-31G*. Все дальнейшие расчеты структур и энергетики процессов комплексообразования с участием ТРТ будут проводиться в дальнейшем с использованием зарядов, рассчитанных методом МК.
Таблица 1 – Значения парциальных зарядов на атомах молекулы топотекана по результатам расчетов в программе Gaussian 03W
| Метод расчета | |||||||||
| HF | DFT | ||||||||
| Базис расчета | Базис расчета | ||||||||
| STO-3G | 3-21G | 6-31G | 6-31G* | 6-31G** | STO-3G | 3-21G | 6-31G | 6-31G* | 6-31G** |
| -0.060483 | -0.061059 | -0.041854 | -0.028568 | -0.040071 | -0.086297 | -0.058345 | -0.041784 | -0.031461 | -0.039251 |
| -0.206055 | -0.180555 | -0.075881 | -0.091352 | -0.096727 | -0.247898 | -0.179270 | -0.083255 | -0.099537 | -0.097865 |
| -0.206055 | 0.294707 | 0.310239 | 0.368242 | 0.365337 | 0.249193 | 0.215341 | 0.224060 | 0.275042 | 0.265238 |
| 0.279377 | -0.721162 | -0.791642 | -0.703419 | -0.695902 | -0.488088 | -0.626277 | -0.695302 | -0.622087 | -0.611229 |
| 0.279377 | 0.135714 | 0.161424 | 0.121076 | 0.121729 | -0.049682 | 0.065928 | 0.086920 | 0.053193 | 0.059737 |
| -0.526421 | 0.945673 | 1.032012 | 0.799679 | 0.800911 | 0.548599 | 0.761131 | 0.857194 | 0.671946 | 0.674067 |
| 0.001011 | -0.425548 | -0.427577 | -0.355091 | -0.354852 | -0.112114 | -0.297733 | -0.298731 | -0.238365 | -0.243017 |
| 0.687908 | -0.342761 | -0.294552 | -0.307350 | -0.307835 | -0.079101 | -0.234523 | -0.173845 | -0.187331 | -0.188365 |
| -0.166502 | -0.642865 | -0.706116 | -0.620744 | -0.621190 | -0.386843 | -0.532092 | -0.599335 | -0.531613 | -0.531087 |
| -0.100061 | -0.598224 | -0.675657 | -0.503867 | -0.503895 | -0.321068 | -0.473215 | -0.552770 | -0.412662 | 0.414556 |
| -0.447736 | 0.831662 | 0.875144 | 0.743426 | 0.740615 | 0.343712 | 0.592873 | 0.639178 | 0.541338 | 0.541991 |
| -0.428335 | 0.316332 | 0.420915 | 0.314743 | 0.309232 | 0.092891 | 0.252019 | 0.358764 | 0.260313 | 0.263284 |
| 0.493331 | 0.095700 | 0.070278 | 0.090906 | 0.089821 | -0.121881 | 0.006555 | -0.026286 | -0.002035 | -0.000858 |
| 0.166298 | -0.660724 | -0.738078 | -0.655044 | -0.654577 | -0.366263 | -0.536708 | -0.618953 | -0.558112 | -0.557706 |
| -0.077616 | -0.368422 | -0.382536 | -0.315701 | -0.309527 | -0.018394 | -0.209503 | -0.214063 | -0.166762 | -0.167652 |
| -0.444477 | 0.444802 | 0.516122 | 0.434119 | 0.434409 | 0.412177 | 0.407540 | 0.485881 | 0.408689 | 0.410286 |
| -0.121480 | 0.105450 | 0.171231 | 0.163407 | 0.148126 | -0.145520 | 0.024096 | 0.080061 | 0.065248 | 0.061545 |
| 0.448491 | -0.131435 | -0.138480 | -0.104875 | -0.101666 | -0.033598 | -0.095919 | -0.101617 | -0.064376 | -0.066399 |
| -0.080805 | -0.616363 | -0.717901 | -0.638095 | -0.639543 | -0.530996 | -0.569293 | -0.673067 | -0.600407 | -0.601866 |
| -0.041577 | -0.301902 | -0.273094 | -0.282336 | -0.283298 | -0.153684 | -0.271651 | -0.230568 | -0.245388 | -0.241871 |
| -0.557146 | 0.380841 | 0.436406 | 0.378978 | 0.378433 | 0.332206 | 0.371577 | 0.428952 | 0.376469 | 0.376416 |
| -0.131352 | 0.211807 | 0.223788 | 0.217388 | 0.217818 | 0.061843 | 0.160457 | 0.160424 | 0.162011 | 0.160385 |
| 0.340930 | -0.229542 | -0.218849 | -0.205075 | -0.203501 | -0.151034 | -0.233893 | -0.215042 | -0.203394 | -0.200448 |
| 0.076088 | -0.634793 | -0.666862 | -0.599049 | -0.597483 | -0.343934 | -0.514892 | -0.536784 | -0.476458 | -0.475002 |
| -0.381039 | -0.350671 | -0.330981 | -0.294683 | -0.293920 | -0.194330 | -0.279282 | -0.254033 | -0.225746 | -0.223514 |
| -0.202298 | 0.670953 | 0.733412 | 0.604184 | 0.599613 | 0.404378 | 0.534051 | 0.595727 | 0.490010 | 0.484949 |
| -0.202298 | 0.613203 | 0.707199 | 0.651952 | 0.647461 | 0.264855 | 0.508933 | 0.607603 | 0.542834 | 0.546180 |
| 0.480375 | -0.527321 | -0.594899 | -0.499048 | -0.495420 | -0.236597 | -0.451647 | -0.534526 | -0.444006 | -0.450138 |
| 0.351288 | -0.277913 | -0.205406 | -0.229116 | -0.234592 | -0.356955 | -0.262513 | -0.170500 | -0.198110 | -0.188582 |
После полученных значений парциальных зарядов на атомах молекулы топотекана, можем построить графическую модель молекулы в программе ChemCraft 1.4 (см. рисунок 2.1).
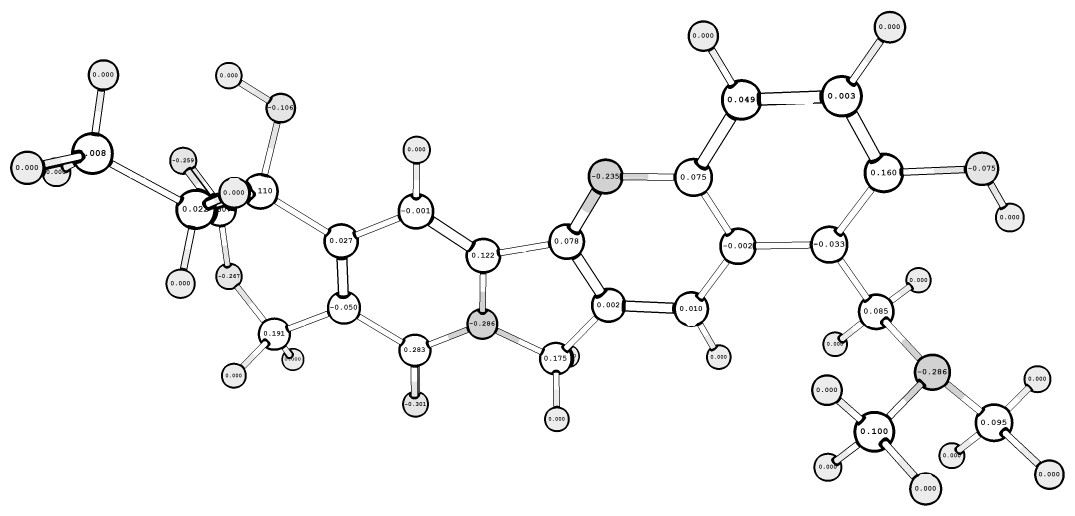
а)

б)
Рисунок 2.1 – Пространственные структуры и значение парциальных атомных зарядов на молекуле топотекана по результатам расчетов:
а) HF_STO-3G и б) DFT_6-31G**
Дата добавления: 2015-09-11; просмотров: 136 | Поможем написать вашу работу | Нарушение авторских прав |