В качестве О—Н-кислот могут выступать любые соединения типа R—О—Н Их диссоциация, без указания основания, к которому переходит протон, может быть представлена следующей схемой:
R—0—Н  R—0:- + Н+
R—0:- + Н+
R—О—Н является тем более сильной кислотой, чем больше электроотрицательность заместителя R и чем сильнее выражен его –М -характер. Наиболее простая О–Н-кислота, несомненно, вода. Известно, что вода относительно слабая кислота (рКа = 15.74, исходя из рКb = 14.00 и [Н2О] = 55.5 для чистой воды). Поэтому вещества, кислотность которых соизмерима с кислотностью воды или меньше ее, в водной среде практически не проявляют своих кислотных свойств. Очень слабые кислоты, для которых рКа больше, чем ~ 16, в водной среде не проявляют себя как кислоты, поскольку концентрация H3O+, которую они создают в воде, оказывается меньше, чем концентрация Н3О+, создаваемая при автопротолизе самой воды:
H2O+H2O H3O++ ОН‑
Вследствие этого относительные кислотности (рКа) слабых кислот совсем не могут быть определены в воде. Если кислоты достаточно сильны (довольно низкие рКа), они практически полностью ионизируются в воде и будут, по-видимому, иметь одинаковую силу, например НС1, HNO3, НС1О4 и т. д. Это явление называют выравнивающим эффектом воды.
Область сравнительного определения рКа может быть расширена при использовании в качестве растворителя как более сильных, так и более слабых оснований, чем Н2О. Проводя соответствующие измерения в ряде растворителей с увеличивающейся основностью (и используя в качестве эталона кислоту, чья кислотность находится почти у нижнего предела значений кислотности в одном растворителе и у почти верхнего предела — в другом растворителе), можно определить силу кислоты вплоть до таких слабых кислот, как метан (pKa ~ 43). Для приобретения кислых свойств молекула О—Н-кислоты должна иметь достаточно электроотрицательный заместитель R. Наибольший эффект достигается, если этот заместитель обладает к тому же еще –М-характером. Рассмотрим теперь кислотность О–Н -кислот по конкретным классам соединений.
Спирты. В одноатомных насыщенных спиртах роль заместителей R играют различные алкильные радикалы. Поскольку эти заместители менее электроотрицательны, чем водород, то наиболее типичные спирты не должны быть более сильными кислотами, чем вода. И действительно, их кислотные свойства в разбавленных водных растворах не проявляются, они не титруются щелочью и т. д. Тем не менее спирты представляют собой типичные кислоты и способны, например, к растворению щелочных и щелочноземельных металлов с выделением водорода
R—0—H + Na (K, Ca и т. д.)  R—0:- +Na+ (К+, Са+ и т. д) + 0, 5Н2
R—0:- +Na+ (К+, Са+ и т. д) + 0, 5Н2
В результате образуются алкоголяты соответствующих металлов, которые можно рассматривать либо как соли спиртов, либо как аналоги щелочей алкоголятные анионы R—0:– обладают сильными основными свойствами, поэтому могут существовать только в среде соответствующего безводного (абсолютного) спирта, так как добавление сколько-нибудь заметных количеств воды сдвигает вправо равновесие:
R–0:- + Н2О R–0–Н +:ОН–
Следовательно, в растворе спирта вода является более сильной кислотой, чем спирт, а гидроксильный анион — более слабым основанием, чем алкоголят-анион. Введение в молекулу спирта электроотрицательных заместителей существенно увеличивает кислотность замещенных спиртов, что видно из следующих примеров:
Спирт рКа
Метанол СН3ОН 15.5
2-Хлорэтанол ClCH2CH2OH 14.3
2, 2, 2-Трихлороэтанол Cl3CCH2OH 12.2
2, 2, 2-Трифтороэтанол F3CCH2OH 12.2
1, 1, 1, 3, 3, 3-Гексафторо-2-пропанол (СF3)2СНОН 9.3
Нанофторо-2-метил-2-пропанол (СF3)3СОН 5.4
Поскольку гидроксильная группа принадлежит к числу электроотрицательных заместителей, то многоатомные спирты – более сильные кислоты, чем одноатомные, причем их кислотность растет с увеличением атомности и уменьшением расстояний между гидроксильными группами. Относительно более сильными кислотами являются гидратные формы альдегидов и кетонов и ортокислоты, так как в них несколько гидроксильных групп связано с одним и тем же атомом углерода.
Енолы. Поскольку в енолах —С=С–O–Н с реакционным центром связана p-электронная система, то сопряженное основание — енолят-ион—существенно стабилизован за счет полярного резонанса:

Поэтому енолы существенно более сильные кислоты, чем насыщенные одноатомные спирты. Этому способствует также индукционное влияние ненасыщенного заместителя, обладающего электроотрицательным характером. Если двойная или тройная связь в ненасыщенном спирте изолирована от гидроксильной группы хотя бы одним атомом углерода, то резонансная стабилизация аниона отсутствует и остается только индукционное влияние электроотрицательной двойной или тройной связи. Поскольку эффективная электроотрицательность тройной связи больше, чем двойной связи, то пропаргиловый спирт (СНºС—СН3ОН) обладает более кислыми свойствами, чем аллиловый спирт (СН2=СН–СН2ОН).
Фенолы. Сопряженные основания фенолов (фенолят-ионы) в существенной мере стабилизированы за счет полярного сопряжения отрицательно заряженного кислорода с ароматическим циклом:

Делокализация отрицательного заряда возможна и в недиссоциированной молекуле фенола, однако, учитывая разделение заряда, она менее эффективна, чем в анионе. Это ведет к некоторому снижению стремления аниона связываться с протоном. Поэтому фенолы – значительно более сильные кислоты (для самого фенола рКа = 9, 95), чем насыщенные спирты, и проявляют свои кислотные свойства также и в водных растворах. Еще больше кислотность таких фенолов, у которых в ароматическое ядро введены электроотрицательные заместители, особенно –М-заместители в о- и п-положениях, способные к полярному сопряжению с реакционным центром в конечном состоянии:

Одновременно следует учесть, что эффективная электроотрицательность замещенного фенила или любого другого ароматического цикла зависит не только от индукционного влияния соединенных с ним заместителей, а также от полярного резонанса этих заместителей с ароматическим циклом. При этом для заместителей ОR и NR; где R — это алкил или Н, влияние + М -эффекта заместителя на п- и о-положения превышает их индукционное влияние, а для галогенов дело обстоит наоборот:
 рКа 10.00,
рКа 10.00,  7.15
7.15
 10.07
10.07  10.68
10.68
 9.02
9.02 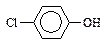 9.38
9.38
 8.39
8.39  5.42
5.42
Карбоксильные кислоты. В карбоксильной группе –С(О)–О–Н с гидроксилом непосредственно соединена карбонильная группа, в достаточной мере электроотрицательная и обладающая сильным – М -характером. Следовательно, как индукционное влияние ацильного радикала R—С(О)—, так и сильный полярный резонанс в карбоксилатном анионе, приводящий к выравниванию зарядов на обоих атомах кислорода (сильная делокализация заряда), обусловливают скачкообразное (более чем на 10 порядков) увеличение кислотности по сравнению с насыщенными спиртами:

Поэтому карбоксильные кислоты целесообразно рассматривать как особый класс кислот со строением X–СООН (результат замещенияR вR—ОН на X—C=О), принимая в качестве кислотного реакционного центра карбоксильную группу в целом и анализируя влияние на кислотность заместителя X. Индукционное влияние эффективной электроотрицательности этого заместителя приводит к увеличению кислотности, хотя это влияние и меньше, чем в случае непосредственной связи электроотрицательного заместителя с гидроксильной группой. Следует иметь в виду, что –М-свойства карбоксилатной группы СО2– существенно слабее, чем у карбоксильной группы, из-за наличия у СО2– отрицательного заряда, препятствующего проявлению акцепторных свойств. Поэтому, если заместитель R обладает +М-xaрактером, исходное состояние оказывается стабилизированным за счет полярного резонанса, отсутствующего в конечном состоянии:



Это приводит к существенному уменьшению силы кислоты, например карбаминовой и угольной кислот, кислых эфиров угольной кислоты и N-замещенных карбаминовых кислот.
Аналогично, если карбоксильная группа непосредственно связана с электронной системой (a, b-ненасыщенные и ароматические карбоксильные кислоты), то полярное сопряжение карбоксильной группы с p-электронной системой выступает в качестве фактора, стабилизирующего исходное состояние, а следовательно, понижающего кислотность. Однако индукционное влияние электроотрицательных p-электронных систем оказывает противоположное действие, будучи при этом более интенсивным. Поэтому такие кислоты все же несколько более сильные, чем их насыщенные аналоги.
Как и следовало ожидать, замещение негидроксильного атома водорода в молекуле муравьиной кислоты на алкильную группу приводит к образованию более слабой уксусной кислоты, поскольку электронодонорный индуктивный эффект алкильной группы понижает остаточное сродство к электрону атома кислорода, связанного с протонизируемым водородом, и, следовательно, уменьшает силу кислоты. В алкилзамещенном анионе повышенная электронная плотность на атоме кислорода способствует дестабилизации аниона и его рекомбинации с протоном:

Действительно, для уксусной кислоты рКа = 4.76, а для муравьиной кислоты рКа = 3.77. Однако степень структурного изменения, проявляющегося в такой небольшой молекуле, как муравьиная кислота, при замене атома водорода на метильную группу, настолько велика, что приведенное выше простое объяснение может и не отражать основные причины наблюдаемого различия. Вполне возможно, что различная способность к сольватации в случае этих двух кислот объясняется заметным различием форм их молекул, а также различным распределением заряда. Следует напомнить, что константа кислотности рКа связана с изменением стандартной свободной энергии ионизации DG° уравнением:
DG° = 2.303 RT ×lg Ka
ичтоDG° включаеткакэнтальпийный,такиэнтропийный члены:
DG°= DH °- Т DS°,
Для ионизации уксусной кислоты в воде при 25°С (рКа =1.79×10-5) = 27.2 кДж (6.5 ккал), DH °= –0.5 кДж (–0.13 ккал) и DS ° = –92 Дж/град (–22 кал/град) [т. е. TDS o = –27.6 кДж (–6.6 ккал)], тогда как для муравьиной кислоты (рКа = 17.6×10-5), DG° = 21 кДж (5.1 ккал), DH° = –0.3 кДж (–0.07 ккал), DS ° = –74 Дж/град (–18 кал/град) [т. е. TDS = –21.3 кДж 5.17 ккал)]. Столь малые значения DH ° почти полностью обусловлены тем фактом, что энергия, необходимая для диссоциации связи О—Н в недиссоциированных карбоновых кислотах, компенсируется энергией, выделяемой при сольватации образующихся ионов.
Таким образом, различие в значениях DG ° для муравьиной и уксусной кислот, обусловливающее различие их Ка, является результатом различия их энтропийных (DS o) членов. Поскольку в каждом равновесии участвуют по два типа частиц с одной и другой стороны, различия в энтропии перехода при диссоциации будут невелики. Однако по одну сторону равновесия обе частицы являются нейтральными молекулами, а по другую — ионами. Поэтому основной вклад в величину DS ° вносит, судя по всему, большая степень упорядоченности сольватных оболочек молекул воды, окружающих ионы RCO2– и Н3О+ по сравнению со степенью упорядоченности этих молекул в самой воде. Увеличение упорядоченности, однако, совсем не так велико, как можно было бы ожидать, поскольку в самой воде упорядоченность уже достаточно велика. Таким образом, различие в силе метановой и этановой кислот в действительности надо связывать с различной сольватацией анионов этих кислот, как это и предполагалось выше.
Дальнейшее введение алкильных групп в уксусную кислоту оказывает гораздо меньший эффект. Этот эффект является по существу эффектом второго порядка, и его влияние на силу кислоты не всегда можно предсказать, поскольку определенную роль могут играть стерические и другие факторы. Значения рКа для ряда гомологов этановой кислоты приведены ниже:
| рКа | рКа | ||
| СН3С02Н | 4.76 | Ме3ССО2Н | 5.05 |
| МеСН2СО2Н | 4.88 | Ме(СН2)2СО2Н | 4.82 |
| Ме2СНСО2Н | 4.86 | Ме(СН2)3СО2Н | 4.86 |
Если рядом с карбоксильной группой находится атом углерода, имеющий двойную связь, сила кислоты возрастает. Так, для акриловой (пропеновой) кислоты, СН2=СНСО2Н, рКа = 4.25, тогда как для ее насыщенного аналога — пропановой кислоты — рКа = 4.88. Это связано с тем, что у ненасыщенного a-атома углерода, находящегося в sр2-гибридизованном состоянии, из-за относительно большего вклада s-орбиталей электроны оттянуты несколько ближе к ядру, чем у насыщенного атома углерода, находящегося в sр3-гибридизованном состоянии. Вследствие этого sр2-гибридизованные атомы углерода обладают меньшей способностью отдавать электроны, чем насыщенные sр3- гибридизованные атомы; именно поэтому пропеновая кислота, уступающая по силе метановой, все же несколько сильнее пропановой. Этот эффект еще больше выражен для sp-гибридизованного атома углерода тройной связи; так, значение рКа пропиновой (пропиоловой) кислоты СНºССО2Н равно 1.84. Аналогичная ситуация наблюдается и для атомов водорода в этене и этине; в этене атомы водорода имеют несколько более выраженные кислотные свойства, чем в этане, в то время как в этине атомы водорода являются настолько кислыми, что легко замещаются некоторыми металлами.
Еще более заметное влияние оказывает введение в молекулу алифатических кислот электроноакцепторных заместителей. Так, введение галогена, индуктивный эффект которого действует в направлении, противоположном индуктивному эффекту алкильной группы, должно увеличивать силу галогензамещенной кислоты, что действительно и наблюдается, как это видно из приведенных ниже значений ряда кислот:
| Кислота | рКа | Кислота | рКа |
| СН3–СO2Н | 4.76 | JСН2–СО2Н | 3.16 |
| FCH2-CO2H | 2.57 | (Cl)2СН–СO2Н | 1.25 |
| С1СН2-СО2Н | 2.86 | (Cl)3С–СО2Н | 0.65 |
| BrCH2–CO2H | 2.90 |
Относительное влияние различных галогенов соответствует ожидаемому: фтор является наиболее электроотрицательным (электроноакцепторным), и сила фторэтановой кислоты в 100 раз выше силы этановой кислоты. Индуктивный эффект атомов галогенов гораздо выше индуктивного эффекта, вызываемого (в противоположном направлении) введением алкильных групп, и дальнейшее введение атомов галогена в молекулу еще более усиливает кислотность; так, трихлорэтановая кислота — уже очень сильная кислота.
Важно напомнить, что Ка (а следовательно, и рКа) связана с DG ° ионизации и что DG °включает как DH °, так и DS °. Было показано, что в ряду галогензамещенных этановых кислот DH °при переходе от одного соединения к другому изменяется незначительно. Наблюдаемое в этом ряду изменение DG ° определяется главным образом различие в значениях DS °, которое в свою очередь обусловлено влиянием атома галогена на делокализацию отрицательного заряда во всем анионе:

В анионе галогензамещенной кислоты отрицательный заряд распределен более равномерно по сравнению с незамещенным этаноат-анионом, заряд которого сконцентрирован в основном на группе СО2–. Это приводит к тому, что введение атома галогена облегчает возможность окружения иона молекулами воды, т. е. его сольватацию. Таким образом, ионизация галогензамещенных этановых кислот сопровождается меньшим снижением энтропии по сравнению с самой этановой кислотой. Этот эффект особенно сильно проявляется в случае CF3CO2H (рКа = 0.23), для которой DG ° = 1.3 кДж (0.3 ккал), тогда как для СН3СО2Н DG °= 27.2 кДж (6.5 ккал); в то же время значения DН ° для обеих кислот мало отличаются друг от друга.
Введение атома галогена в положение, более удаленное от карбоксильной группы, чем a-положение, оказывает гораздо меньшее влияние. Это объясняется тем, что индуктивный эффект быстро ослабевает при передаче вдоль насыщенной цепи, в результате чего отрицательный заряд становится все менее «размытым», оставаясь концентрированным в карбоксилат-анионе. В этом случае кислота все больше напоминает соответствующую незамещенную алифатическую кислоту, как это можно видеть из приведенных в табл. 2 значений рКа.
Другие электроноакцепторные группы, например NR3, CN, NO2, SO2R, CO, CO2R, увеличивают силу простых алифатических кислот. Аналогичное влияние оказывают гидроксильные и метоксильные группы. Неподеленные электроны атомов кислорода групп ОН и ОМе не способны проявлять мезомерный эффект в направлении, противоположном индуктивному эффекту, из-за наличия в молекуле насыщенного атома углерода. Значения рКа соответствующих кислот приведены ниже:
Таблица 2
Значения рКа ряда карбоновых кислот
| Кислота | рКа | Кислота | рКа |
| НСООН | 3.75 | O2NCH2CO2H | 1.68 |
| СН3СООН | 4.76 | Me3NCH2CO2H | 1.83 |
| CICH2COOH | 2.86 | NCCH2CO2H | 2.47 |
| CI3C—COOH | –0.4 | ЕtO2ССН2СO2Н | 3.35 |
| CI(CH2)2COOH | 4.08 | МеОСН2СО2Н | 3.53 |
| CI(CH2)3COOH | 4.52 | НОСН2СООН | 3.83 |
| CH3СН2СН2СO2Н | 4.82 | МеСОСН2СО2Н | 3.58 |
| CH3СН2СНС1СО2Н | 2.84 | CH2=CH–COOH | 4.56 |
| CH3СНС1СН2СО2Н | 4.06 | CHºCH–COOH | 1.84 |
Особого внимания заслуживает рассмотрение ароматических карбоновых кислот. Бензойная кислота (рКа = 4.20) является более сильной кислотой, чем ее насыщенный аналог — циклогексанкарбоновая кислота (рКа = 4.87). Это дает основание предположить, что фенильная группа, подобно двойной связи, является менее электронодонорной (по сравнению с насыщенной системой углеродных атомов) по отношению к карбоксильной группе из-за наличия sр2-гибридизованного атома углерода, присоединенного к карбоксильной группе. Введениеэлектронодонорных алкильных групп в ароматическое кольцо очень слабо влияет на силу образующейся кислоты (ср. с влиянием таких групп в фенолах), в то время как введение аминных групп заметно уменьшает кислотность:
рКа
С6Н5СО2Н 4.20
м-МеС6Н4СО2Н 4.24
п-МеС6Н4СО2Н 4.34
м-Н2NС6Н4СО2Н 4.74
п-Н2NС6Н4СО2Н 4.85
 В замещенных бензойных кислотах электроотрицательные заместители приводят к увеличению кислотности в соответствии с комбинированным влиянием их индукционного эффекта и полярного сопряжения с ароматическим циклом. Так, при введении электроноакцепторных групп, например группы NO2, в пара -положение кислотность увеличивается.
В замещенных бензойных кислотах электроотрицательные заместители приводят к увеличению кислотности в соответствии с комбинированным влиянием их индукционного эффекта и полярного сопряжения с ароматическим циклом. Так, при введении электроноакцепторных групп, например группы NO2, в пара -положение кислотность увеличивается.
| рКа | |
| С6Н5С02Н | 4.20 |
| o-O2NC6H4CO2H | 2.17 |
| м-O2NC6H4CO2H | 3.45 |
| п-O2NC6H4CO2H | 3.43 |
| 3,5-(O2N)2C6H3CO2H | 2.83 |
Иначе обстоит дело в случае о-замещенных бензойных кислот, так как они оказываются более сильными кислотами, чем незамещенная бензойная кислота, независимо от природы заместителя:

 рКа 4.18 2.94
рКа 4.18 2.94

 3.92 2.17
3.92 2.17
 3.00
3.00
Такая «аномалия» характерна и для ряда других реакций производных о-замещенных фенилов. Она получила название орто-эффект. Особенно заметное влияние NО2-группы в орто -положении обусловлено, по-видимому, тем, что на очень коротком расстоянии индуктивный эффект действует сильнее всего, но нельзя исключать и прямое взаимодействие между соседними группами NO2 и СО2Н. В ряде случаев орто-эффект вызван дополнительным влиянием стерического фактора и внутримолекулярной водородной связью, но одними этими факторами невозможно удовлетворительно объяснить все случаи проявления орто-эффекта, в том числе и для о-замещенных бензойных кислот. Существует мнение, что важную роль играют здесь особые эффекты специфической сольватации.
Наличие таких групп, как ОН, ОМе или галоген, обладающих электроноакцепторным индуктивным эффектом, но электронодонорным мезомерным эффектом в орто- или пара-положениях, может привести к тому, что пара-замещенные кислоты окажутся слабее мета-замещенных, а иногда и даже слабее исходной незамещенной кислоты, как, например, в случае пара-гидроксибензойной кислоты:

рКа ХС6Н5СО2Нпри различных Х:
| Н | С1 | Вг | ОМе | ОН | |
| о -Изомер | 4.20 | 2.94 | 2.85 | 4.09 | 2.98 |
| м -Изомер | 4.20 | 3.83 | 3.81 | 4.09 | 4.08 |
| п -Изомер | 4.20 | 3.99 | 4.00 | 4.47 | 4.58 |
Следует отметить, что этот компенсирующий эффект становится более выраженным при переходе: С1» Вг ОН, т.е. при возрастании способности атома, связанного с ароматическим кольцом, отделяться вместе со своей электронной парой. Важно подчеркнуть, что здесь, как и в приведенных выше случаях, возможно, сказывается влияние различного распределения заряда в анионах на их сольватацию, т. е. на значение TDS °, связанное со степенью упорядоченности молекул растворителя в сольватной оболочке, что может стать причиной наблюдаемых различий в значениях рКа.
Поведение о-замещенных кислот, как отмечалось выше, часто аномально. Иногда они оказываются более сильными, чем можно было ожидать, исходя из предположения о прямом взаимодействии соседних групп. Так, внутримолекулярные водородные связи стабилизируют анион о-гидроксибензойной (салициловой) кислоты за счет делокализации его заряда, что нехарактерно ни для м- и п-изомеров, ни для о-метоксибензойной кислоты.

Внутримолекулярные водородные связи могут, конечно, действовать в молекуле недиссоциированной кислоты так же, как и в ее анионе, но, вероятно, в последнем случае более эффективно, чем в первом (с соответствующей относительной стабилизацией), так как отрицательный заряд на атоме кислорода в анионе способствует образованию более сильной водородной связи. Этот эффект даже более выражен, когда водородные связи образуются с гидроксильными группами в обоих орто-положениях; так, для 2,6-дигидроксибензойной кислоты рКа = 1.30.
Дикарбоновые кислоты. Простую интерпретацию невозможно дать и константам диссоциации двухосновных карбоксильных кислот, диссоциирующих в две стадии

Значения величин К2 кислот (HO2C[CH2]nCO2H) явно «аномальны»:
Значение n рКа (2)
0 3.97
1 5.40
2 5.34
3 4.97
Для сравнения CH3COOH 4.76
Повышенная кислотность двухосновных кислот объясняется электроноакцепторным характером второй карбоксильной группы. Однако ее влияние быстро уменьшается по мере того, как карбоксильные группы оказываются разделенными более чем одним насыщенным атомом углерода.
При сопоставлении кислотности двухосновных и одноосновных кислот для первых необходимо предварительно ввести так называемую статистическую поправку. Она учитывает наличие в недиссоциированной двухосновной кислоте двух центров кислотности, а в ее анионе - двух центров основности, вследствие чего значения К1 в два раза больше, а значения К2 — в два раза меньше, чем этого следовало бы ожидать при идентичном влиянии факторов строения, но при наличии только одного центра кислотности или основности. Благодаря этому для указанных кислот предельное значение К1, достигаемое при увеличении n, на lg2 = 0.30 единиц рКа меньше, а предельное значение рК2 — на ту же величину больше, чем аналогичное предельное значение рКа для кислот СНз(СН2)nСООН:
Кислота рК1 рК2
HOOC(CH2)4COOH 4.42 5.28
HOOC(CH2)5COOH 4.51 5.31
CH3(CH2)5COOH 4.84 —
С учетом статистической поправки значение рК2 щавелевой кислоты (n=0) получается примерно на 0, 8 единицы меньше, а для малоновой кислоты (n=1) — на 0.6 единицы больше рКа пропионовой кислоты. Таким образом, создается впечатление, что при n=0 карбоксилатная группа —С00– является более электроотрицательным заместителем, чем СН3, а при п= 1 ее электроотрицательность существенно меньше, чем у метила. С точки зрения современных теоретических представлений, этому трудно дать удовлетворительное объяснение.
В определенных случаях на кислотность многоосновных карбоксильных кислот заметное влияние оказывает наличие внутримолекулярной водородной связи. Хорошей иллюстрацией являются значения рК1 и рК2 фумаровой (рК1 = 3.02; рК2 =4.38) и малеиновой (рК1 = l.92; рК2 =6.23) кислот. Эти кислоты диссоциируют по следующим схемам:


Наличие внутримолекулярной водородной связи в моноанионе малеиновой кислоты приводит к его дополнительной стабилизации. Так как этот моноанион является конечным состоянием для первой ступени диссоциации и исходным для второй ступени, то стабилизация приводит к соответствующему увеличению K1 и уменьшению К2. Это особенно заметно при сопоставлении K1 и К2 для фумаровой кислоты, в которой внутримолекулярная водородная связь невозможна в результате транс-положения карбоксильных групп. Щавелевая, малоновая и янтарная кислоты уступают по второй ступени диссоциации муравьиной и пропановой кислотам соответственно. Это объясняется тем, что в случае дикарбоновых кислот второй протон приходится удалять из отрицательно заряженных частиц, содержащих электронодонорный заместитель, т. е. группу СО2–, которая, как можно ожидать, дестабилизирует анион по сравнению с недиссоциированной кислотой в большей степени — эффект, который отсутствует при диссоциации незамещенной кислоты:

Сульфиновые и сульфоновые кислоты, фосфоновые и фосфоновые кислоты. В указанных кислотах с гидроксильной группой связаны очень электроотрицательные заместители, обладающие одновременно – М -характером. Благодаря этому как сульфиновые, так и сульфоновые кислоты относятся к числу сильных кислот. Из известных значений рКа в качестве примеров можно привести следующие: для бензенсульфиновой кислоты C6H5SOOH рКа =1.5, а для бензенсульфоновой кислоты C6H5SO2OH рКа = 0.7. Ниже приводятся значения рКа для некоторых фосфиновых и фосфоновых кислот.
Кислота рКа
Фосфиновые кислоты
Диметилфосфиновая кислота 3.08
Бис(трет-бутил)-фосфиновая кислота 4.24
Дифенилфосфиновая кислота 2.10
Фосфоновые кислоты рК1 рК2
Метилфосфоновая кислота 2.38 7.74
Хлорметилфосфоновая кислота 1.40 6.30
Бензенфосфоновая кислота 1.82 7.07
Дата добавления: 2015-09-11; просмотров: 276 | Поможем написать вашу работу | Нарушение авторских прав |